
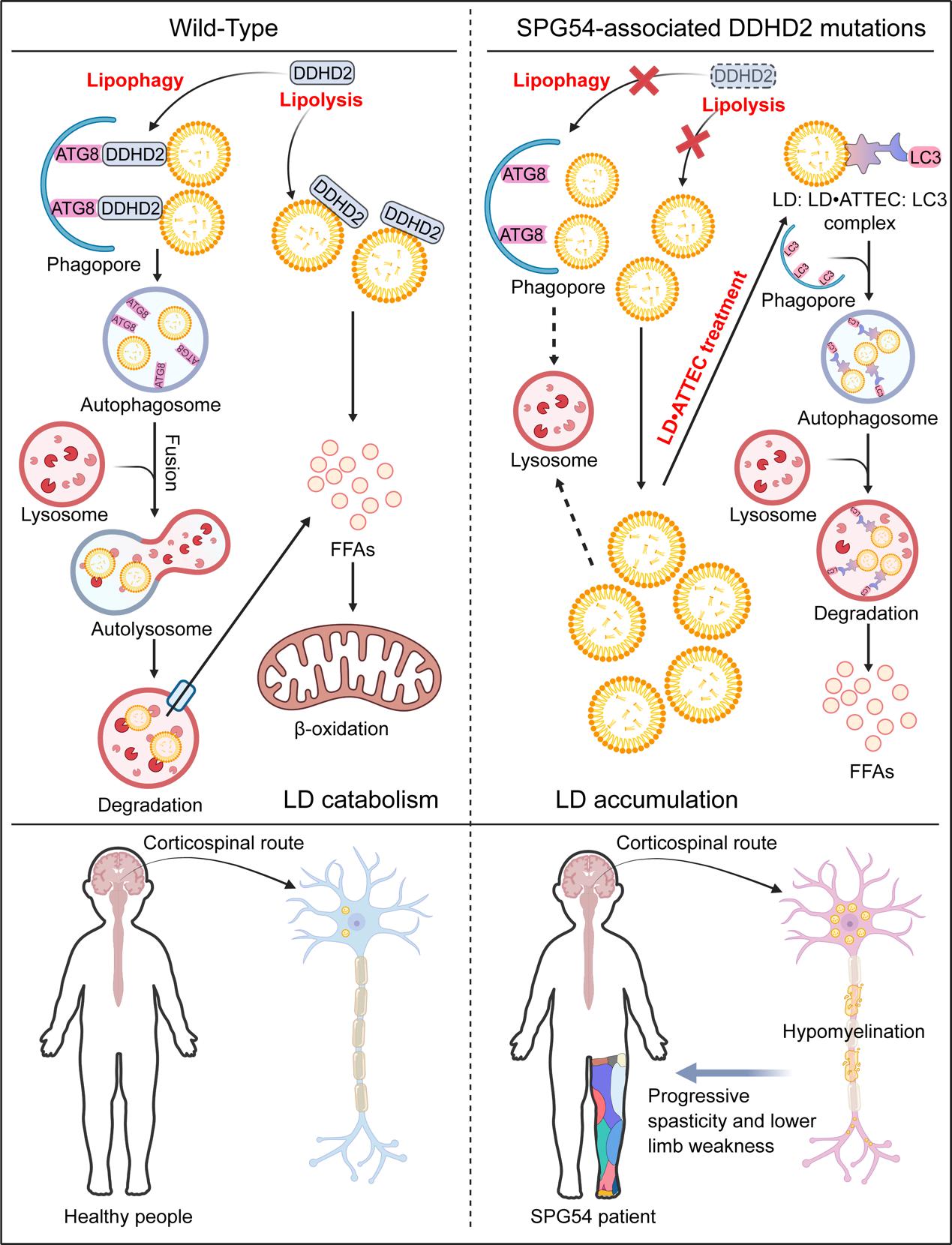
遗传性痉挛性截瘫(Hereditary Spastic paraplegia, HSP)是一组遗传性神经退行性疾病,其特征是下肢进行性痉挛和无力。HSP可以分为单纯型和复杂型,单纯型主要涉及运动神经元退化,而复杂型除了神经学表现外,还会出现其他非神经性症状。目前已经确定了80个与HSP相关的基因或位点,编号为SPG1-80,其遗传模式包括常染色体显性(AD)、常染色体隐性(AR)和X染色体连锁。这些基因涉及各种生物学过程,如轴突运输、内质网形状和动态变化、膜转运、脂质代谢和线粒体功能等。其中一种罕见的复杂型AR HSP是54型痉挛性截瘫(SPG54),由DDHD2基因的双等位基因突变所引起。2大多数DDHD变异会导致蛋白质产物功能丧失,以无义或移码截断突变为主,而一小部分变异为错义突变。 SPG54发病早,通常在生命前十年出现症状,包括痉挛步态、智力残疾、纤维束体变薄以及通过磁共振波谱技术在大脑中检测到脂质峰升高,病理学检查发现SPG54患者大脑神经元中脂滴(lipid droplet,LD)大量积累,提示脂质分解代谢紊乱可能是SPG54的主要致病机理。
DDHD2与DDHD1、SEC23IP共属于细胞内磷脂酶A1(PLA1)家族。已有生化研究表明,DDHD1和DDHD2具有脂酶的潜力,能在体外水解各种(磷)脂质底物,然而其具体的生理功能和内源性底物尚未完全揭示。敲除DDHD2基因会导致小鼠大脑组织甘油三酯(Triacylglycerol, TAG)水平显著升高,而其他组织无此现象,这种升高可能与神经元中LD累积相关,并由此导致类似SPG54的认知和运动功能障碍。此外,生化检测也证明DDHD2是大脑组织中主要的TAG脂酶。这些发现提示,大脑中有一条由DDHD2主导的TAG代谢途径,破坏该途径可能导致神经元中LD大量累积,产生严重的神经退行性症状。然而,目前人们对DDHD2参与脂质代谢的具体分子机制还不十分清楚。
近日,复旦大学生命科学学院王陈继团队、鲁伯埙团队和复旦大学附属妇产科医院赵世民团队合作在Cell Death & Differentiation杂志发表了题为DDHD2, whose mutations cause spastic paraplegia type 54, enhances lipophagy via engaging ATG8 family proteins的研究论文,该研究发现DDHD2可作为货物受体(cargo receptor),通过与ATG8家族蛋白相互作用来促进脂滴自噬(lipophagy)。

在本研究中,研究人员首先通过亲和纯化质谱(AP-MS)分析发现,DDHD2与自噬相关ATG8家族蛋白(LC3、GABARAP)的多个成员存在相互作用,这些成员在自噬中发挥着至关重要的作用。通过氨基酸序列分析、定点突变和免疫共沉淀实验研究者进一步揭示了 DDHD2 蛋白存在两个LIR (LC3 interaction region)基序,负责介导其与 LC3/GABARAP 的结合。在功能实验中,研究者通过CRISPR/Cas9构建DDHD2敲除细胞系,发现DDHD2 缺失会导致LD累积;而在此基础上过表达DDHD2脂酶活性缺失突变体(S351A)或者LIR突变体均只能部分程度上消解LD,双突变则会使其消解LD作用基本丧失。免疫荧光实验表明DDHD2能够促进LD与LC3B、溶酶体之间的共定位。通过GFP-mCherry livedrop报告系统也证实DDHD2敲除会降低细胞内的脂滴自噬通量。综上所述,DDHD2在LD分解代谢中扮演双重角色,一方面通过其脂酶活性消解LD,另一方面通过与ATG8家族蛋白结合促进LD通过自噬途径降解。
目前临床上对SPG54尚无有效的治疗手段。鉴于LD累积是SPG54的致病因素,如能有效消解病理性LD累积则为一种潜在的靶向治疗思路。LD·ATTECs是鲁伯埙团队开发的一类自噬绑定化合物,其通过疏水作用与选择性LD结合,同时与LC3结合,形成LD/TAG–LD·ATTEC–LC3三元复合物,该复合物可在自噬小体形成过程中绑定LD于自噬小体内进而诱导其通过自噬降解(Cell Research, 2021)。在本研究中,研究人员利用多种细胞模型验证了LD·ATTEC能有效地消解 DDHD2 敲除引起的 LD 积累,这为未来SPG54的治疗提供了一个潜在的靶向策略。
复旦大学生命科学学院博士生贾菲为该论文的第一作者,王陈继、鲁伯埙和赵世民为该论文的共同通讯作者。
原文链接:https://www.nature.com/articles/s41418-024-01261-1