
鲁伯埙课题组针对神经退行性病变亨廷顿病的研究取得重要突破,相关文章发表于高端期刊eLife,复旦大学是文章的第一单位及最终通讯单位。文章为神经退行性病变亨廷顿病领域的两个根本性问题的探索提供了重要信息,并可能推广至类似疾病。
神经退行性疾病引起中枢神经系统特定神经元不正常死亡萎缩,导致神经功能严重缺陷,认知或运动功能障碍,是最重要的神经疾病之一。神经退行性疾病中的亨廷顿病(Huntington’s Disease,HD)由于遗传图景清晰,是此类疾病重要的模式疾病。目前此类疾病,包括亨廷顿病,没有任何根本性治疗的方法。
围绕神经退行性疾病有两个关键的科学问题:第一、如何治疗神经退行性疾病?第二、为何此类疾病的神经元死亡存在细胞特异性?也就是说,为何导致疾病的蛋白在各种神经元表达,但是每种疾病中主要只有特定的一类神经元死亡最严重?
亨廷顿病主要由HTT基因突变产生的突变HTT蛋白的细胞毒性导致。尽管变异HTT蛋白广泛表达,亨廷顿病中的神经退行主要发生在大脑纹状体,特别是其中表达多巴胺受体二型(D2)的中间多棘神经元(medium spiny neuron)是最早死亡的一种。鲁伯埙实验室通过遗传学筛选发现富集于D2型中间多棘神经元的孤儿G蛋白偶联受体GPR52可以有效稳定纹状体的突变HTT蛋白,而非大脑皮层的突变HTT蛋白。因此,GPR52可能通过特异性地稳定纹状体神经元的突变HTT蛋白导致纹状体神经元对变异HTT蛋白更敏感,从而使其在亨廷顿病中更容易死亡。类似的机制也可能存在于其他神经退行性疾病。
鲁伯埙课题组进一步揭示了GPR52调控变异HTT蛋白稳定性的机制。此调控过程依赖于GPR52的G蛋白偶联受体活性和下游的第二信使cAMP,但是并不依赖于经典的cAMP感应蛋白PKA,而是依赖于小G蛋白介导的变异HTT的胞内运输。有趣的是,GPR52是一个单外显子基因,完全位于另外一个基因RABGAP1l的一个内含子中,且方向一致。进一步研究发现,RABGAP1l可以通过其对小G蛋白RAB39B的阻断,对抗GPR52的作用,为HTT蛋白提供相互平衡的调控。这是首次发现一个基因和它内含子所包含的基因在功能上直接相互联系。
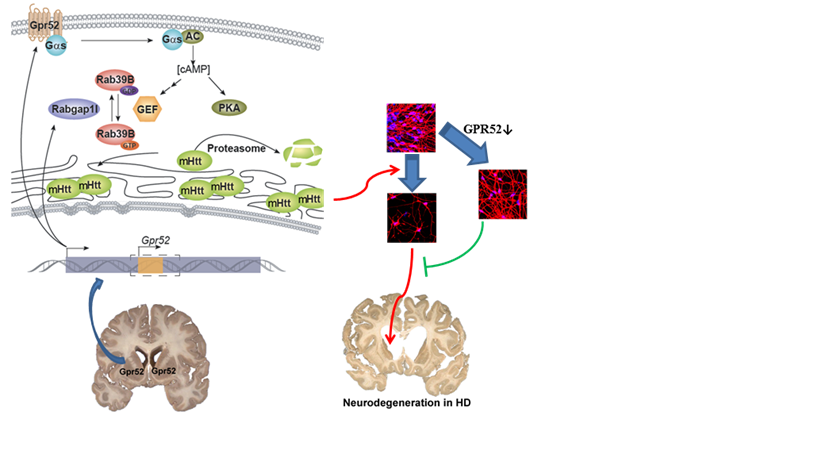
GPR52的发现不仅为疾病神经元死亡的区域特异性提供了可能解释,而且也为疾病治疗提供了可能的药靶。GPR52的敲低或敲除,显著拯救了人类干细胞分化亨廷顿病神经元模型的神经元萎缩及死亡,以及在体果蝇遗传学动物模型的疾病表型。G蛋白偶联受体是目前最主要的一类药物靶点,因此GPR52的制药潜力巨大。后续研究将围绕针对GPR52的药物研发以及哺乳动物模型的验证展开。
鲁伯埙研究员回国两年以来,积极展开神经退行性疾病方面的工作,目前以复旦为第一及通讯单位已发表Nature Neuroscience,eLife, Scientific Reports, FASEB Journal,Trends in Pharmacological Sciences等多篇文章,总影响因子近50。
文章链接:http://elifesciences.org/content/early/2015/03/04/eLife.05449/article-info