
5月5日,生命科学学院李继喜教授团队在程序性细胞坏死研究领域取得重要进展,发现和鉴定了靶向坏死关键蛋白RIP3的新型“刹车”分子TRIM-25;研究论文以《E3连接酶TRIM25泛素化修饰RIP3从而抑制肿瘤坏死因子TNF引起的细胞坏死》(E3 ligase TRIM25 ubiquitinates RIP3 to inhibit TNF induced cell necrosis)为题发表在《细胞死亡与分化》(Cell Death and Differentiation)杂志上。
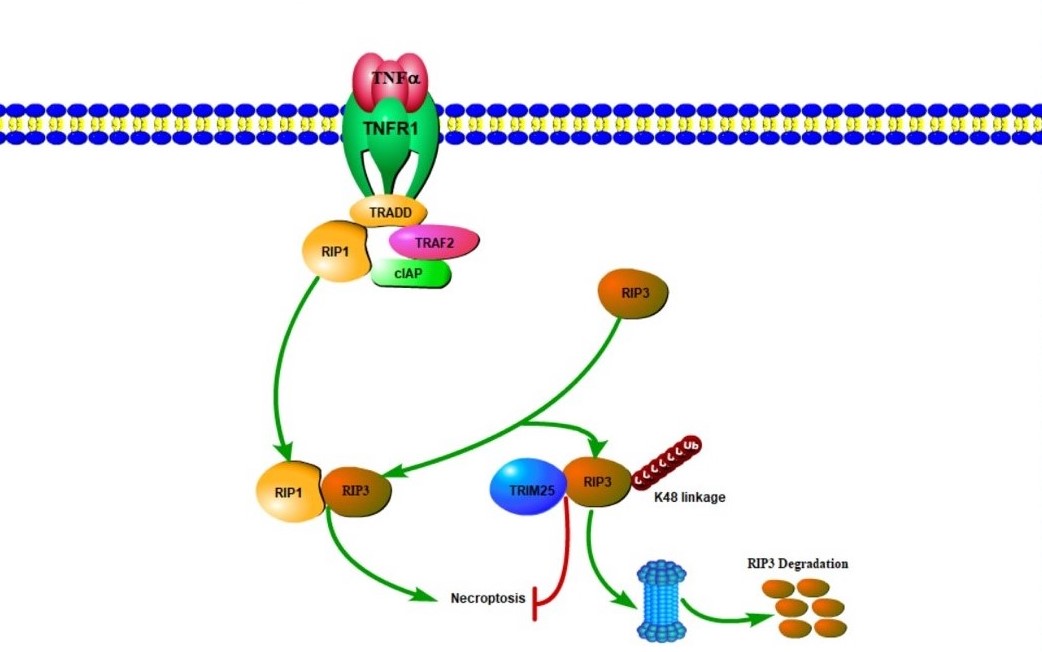
图示:TRIM25介导RIP3的泛素化修饰从而负调控TNF诱导的程序性细胞坏死
程序性细胞坏死与肿瘤发生发展、免疫及代谢性疾病相关,在出血性脑卒中及肝损伤、心肌出血性坏死、神经退行性疾病以及高雪氏病中起着十分重要的作用。程序性细胞坏死可以由多种因素诱导产生,如死亡受体、Toll样受体、胞质内RNA或DNA感知蛋白ZBP1等;而TNF信号引起的细胞坏死主要由坏死小体(necrosome)蛋白RIP1、RIP3和MLKL介导。李继喜课题组长期从事程序性细胞坏死的结构基础与调控机制研究,早期发现RIP1/RIP3介导的坏死小体形成功能性的β淀粉样纤维复合物,作为级联放大平台,激活程序性细胞坏死(2012,Cell);通过结构生物学方法和手段解析了坏死小体的高分辨率三维结构(2018,Cell);发现介导RIP1/RIP3相互作用的RHIM结构域可以跨物种发挥功能,在昆虫固有免疫应答IMD信号通路中激活NF-kB,产生多种抗菌肽(2017,Immunity)。
在本研究中,李继喜团队通过高灵敏质谱方法筛选并鉴定到RIP3的特异性E3泛素连接酶TRIM25。TRIM25与RIP3在体外和体内发生直接相互作用;敲除或者敲低TRIM25基因后细胞死亡显著增强;TRIM25通过K48修饰的多聚泛素化特异性作用于人源RIP3基因的K501氨基酸,进一步通过蛋白酶体途径降解RIP3从而抑制TNF诱导的程序性细胞坏死。这项研究揭示了RIP3的翻译后修饰调控细胞坏死的全新机制,将为开发与细胞坏死相关的药物提供新的作用靶标和治疗方法奠定基础。
博士研究生梅谱成和谢飞艳为论文共同第一作者,李继喜教授为论文通讯作者。生命科学学院丁琛教授、吴家雪教授、王永明教授以及复旦大学附属华山医院陈向军教授给予了支持和帮助。该研究得到了国家自然科学基金委、科技部国家重点研发计划和上海市科委的资助。
全文链接:https://www.nature.com/articles/s41418-021-00790-3