
2022年4月5日,国际学术期刊Cell Reports在线发表了我院胡薇/陆艳团队的研究成果“A chromosome-level genome of the human blood fluke Schistosoma japonicum identifies the genomic basis of host-switching”。该研究运用三代测序、Hi-C、全长转录组(Iso-Seq)等技术完成了首个染色体水平的日本血吸虫参考基因组的组装和注释,并对覆盖整个流行地区的72个基因组进行了重测序,系统解析了日本血吸虫在东亚、东南亚地区的群体遗传结构和分化历史,鉴定了与宿主适应性相关的关键基因,为血吸虫病的防控和药物开发提供了新的思路。
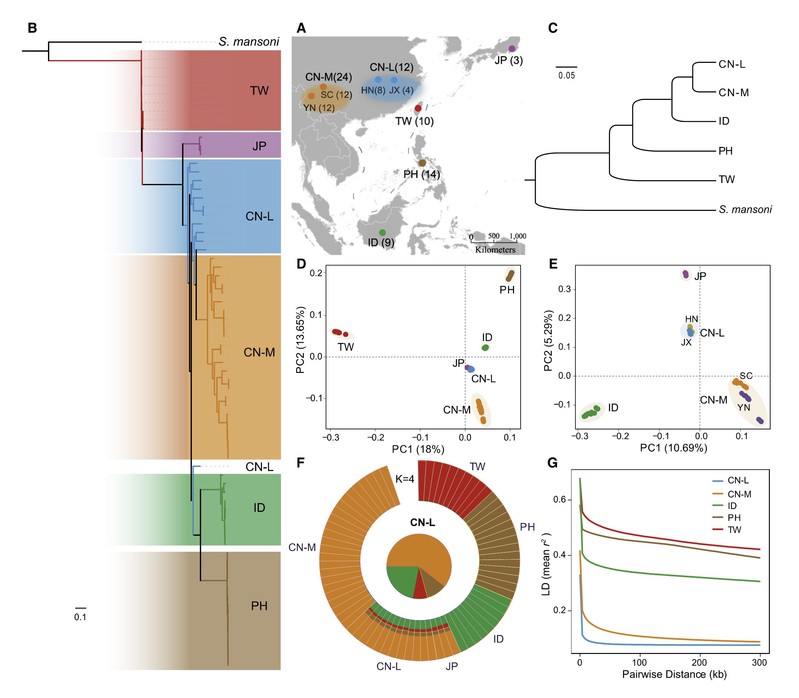
图1 日本血吸虫的样本分布及群体遗传结构
血吸虫病是一种严重威胁人类健康的热带疾病,亚洲流行的日本血吸虫是一种古老的寄生虫,在中国的流行历史已超过2000年。日本血吸虫可感染40种以上的哺乳动物,具有宿主域广且生殖力强的特点。长期以来,由于缺乏高质量的全基因组数据,日本血吸虫的群体结构和适应性进化相关研究受到极大制约。研究团队采集、保藏了亚洲全部6个流行区的72个日本血吸虫样本,通过群体遗传学分析发现日本血吸虫的遗传结构和其地理分布区域一致,在遗传成分上可分为中国大陆、中国台湾、菲律宾和印度尼西亚四个祖源。距今约4.5万年前,亲动物且致病性较弱的中国台湾株率先从其他群体中分离出来,这与中间宿主钉螺在台湾的分离时间相吻合。值得一提的是,与人群有效群体大小的波动一样,日本血吸虫群体在末次冰川期经历了强烈的种群瓶颈效应。
日本血吸虫生活史复杂,需要经历两个宿主寄生阶段才能完成其生命周期。血吸虫对寄生环境即中间宿主及终宿主的适应是其快速流行的必要条件。通过全基因组自然选择信号筛选,研究团队鉴定了一系列与血吸虫的生殖发育和入侵宿主等相关的功能基因,这些基因在不同血吸虫群体之间发生了强烈分化。RNA干扰实验验证了GATAD2A基因与日本血吸虫在终宿主体内的生殖发育和感染维持有关;转录组结果显示Lmln基因在日本血吸虫入侵中间宿主时期(毛蚴)高表达,该基因在中国湖区和中国山区群体中的分化可能与中间宿主钉螺在该地区的相容性差异有关。
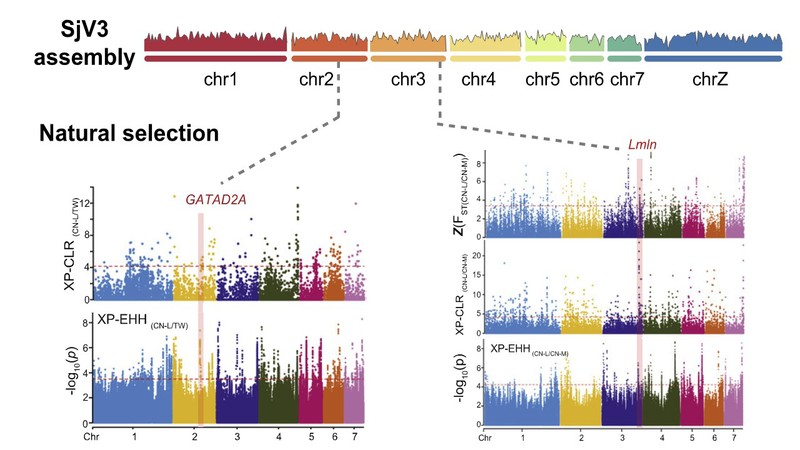
图2 日本血吸虫全基因组受选择信号筛选
该研究提供了迄今为止最全面的日本血吸虫全基因组测序数据,解析了日本血吸虫的适应性进化,鉴定了一系列日本血吸虫在中间宿主和终宿主体内建立维持感染以及生长发育相关的候选基因,并为后续的血吸虫研究提供了重要的数据资源。复旦大学生命科学学院博士后罗芳博士和博士生杨文彬为本文共同第一作者,胡薇教授和陆艳副研究员为该论文的共同通讯作者。复旦大学生命科学学院为第一单位与通讯作者单位。该工作得到了国家自然科学基金委、国家重点研发计划和上海市科委的资助。
全文链接:https://www.cell.com/cell-reports/fulltext/S2211-1247(22)00390-4