
2022年5月30日,复旦大学生命科学学院徐书华团队在Cell Press旗下学术期刊Star Protocols在线发表了题为“A protocol for applying a population-specific reference genome assembly to population genetics and medical studies”的方法技术论文,公布了基于族群特异性参考基因组开展群体遗传学和医学遗传学研究的分析流程(图1)。
构建族群特异的参考基因组的必要性和应用价值已在该团队前期工作中进行了论证(https://life.fudan.edu.cn/99/30/c28140a432432/page.htm)。相比于通用的人类参考基因组,族群特异性参考基因组能够针对特定人群显著提升遗传变异检测的准确度与灵敏性,更高效地揭示群体间的遗传结构与遗传差异,更全面地判别医学疾病相关的低频罕见变异,更精准地辅助遗传诊断。在新一代长读长测序技术(long-read genome sequencing)的助力下,人类泛基因组(pangenome)迎来了黄金发展期,可以预见未来几年内将实现大量不同人群参考基因组的从头组装。因此,迫切需要建立一个具有可操作性、高效性和可重复性的族群特异性参考基因组分析流程为人们探究人类基因组的奥秘提供方法和技术上的便利。
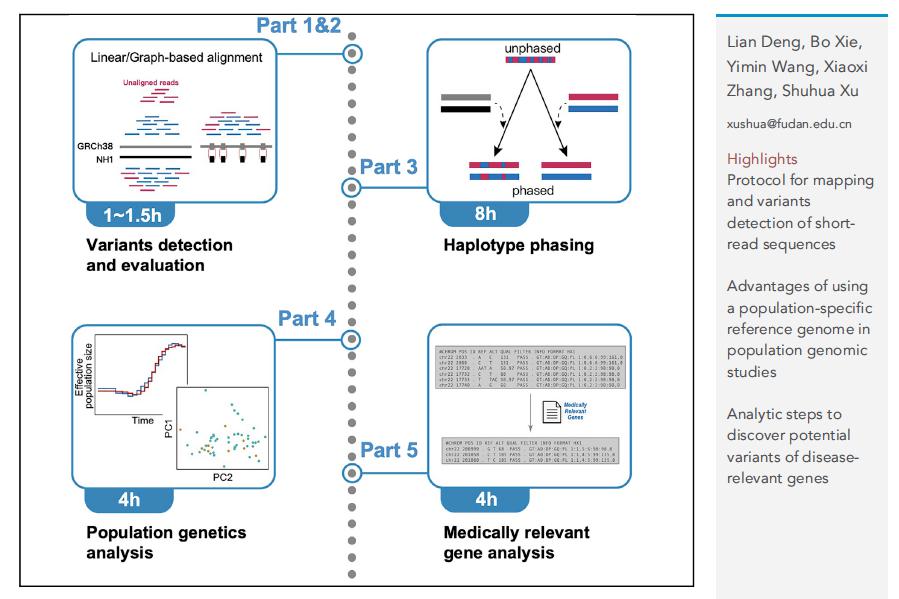
图1 族群特异性参考基因组的应用分析流程图
该分析流程以公共开放数据库中的中国汉族参考基因组22号染色体数据为例,详细展示了1)通过序列线性比对(linear mapping)和构建图基因组(graph genome)的方法检测遗传变异,2)单倍型推断(phasing),3)主成分分析和人群历史推断等常规群体遗传分析,以及4)挖掘潜在的疾病相关基因变异的分析步骤、软件和代码执行命令、文件格式和注意事项等。该流程中大多数分析均采用已公开发布并经历长期实践验证的软件程序包,研究团队对分析过程可能遇到的问题、对应的解决方案以及可替代的软件和方法进行了详细阐述。该流程的发布将有助于发挥族群特异性参考基因组的应用价值和潜力。
复旦大学生命科学学院青年研究员邓恋博士为第一作者,中国科学院上海营养与健康研究所博士生谢波和王亦民以及上海科技大学博士生张晓曦参与了该项工作的完成。复旦大学生命科学学院为第一单位与通讯单位,徐书华教授为本文通讯作者。该项工作获得了国家自然科学基金委、中国科学院先导专项、英国皇家学会牛顿基金、上海市科委等多项基金的资助。
论文链接:https://www.sciencedirect.com/science/article/pii/S2666166722003203