
线粒体在能量代谢和信号转导等生物学过程中发挥着重要作用,是真核细胞不可或缺的细胞器。线粒体DNA耗竭综合征(Mitochondrial DNA Depletion Syndrome,简称MTDPS)是一组罕见的遗传疾病,由参与线粒体DNA(mtDNA)复制和维持的多个基因缺陷所引起,这些缺陷导致mtDNA编码蛋白合成减少和氧化磷酸化(OXPHOS)不足,故而继发各种代谢异常和多系统的异常临床表现。
2023年12月20日,复旦大学生命科学学院王陈继团队和上海市第一妇婴保健院高昆团队应邀在Trends in Molecular Medicine杂志发表了题为Excessive BNIP3/BNIP3L-dependent mitophagy drives FBXL4 mutation-caused mitochondrial DNA depletion disease的综述,概括了MTDPS 13亚型(MTDPS13)致病基因FBXL4突变导致线粒体自噬过度激活的内在分子机制,并讨论了这种致命遗传疾病的潜在治疗策略。
MTDPS13相关症状广泛,个体差异很大,最常见症状包括张力减退、神经发育迟缓和乳酸血症等。具有高能量需求的器官,如大脑、心脏、肌肉和肾系统,通常会受到更严重的影响。患者预后通常较差,平均3岁时死亡率约为30%。MTDPS13致病基因FBXL4是一个Cullin 3家族E3泛素连接酶复合体的底物识别亚基。自2013年首次描述FBXL4基因致病突变以来,在112例常染色体隐性遗传患者中共记录了59种致病突变。与双等位基因缺失突变的患者相比,双等位错义突变、错义/缺失突变复合杂合性患者往往生命更长。这一观察结果意味着错义变体可能在这些个体中发挥亚等位基因效应。
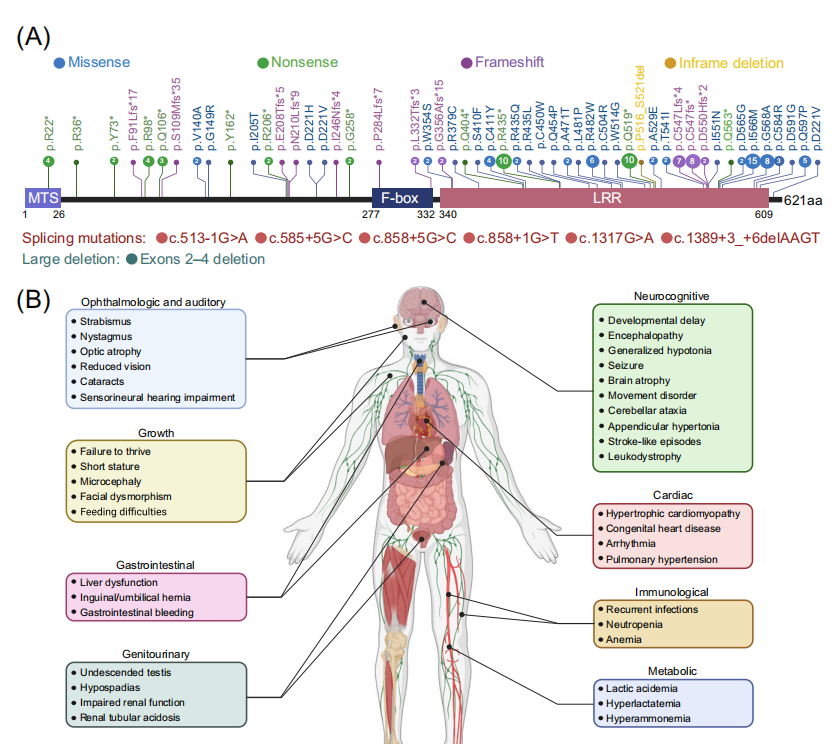
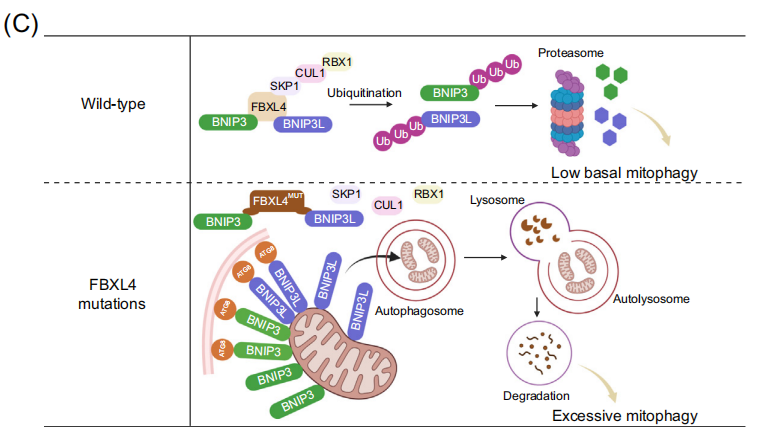
2023年发表的多项研究在该病的分子机理方面取得重大进展,阐明了FBXL4通过控制线粒体外膜自噬受体BNIP3和BNIP3L来抑制基础线粒体自噬(basal mitophagy)的关键作用。线粒体外膜蛋白BNIP3和BNIP3L具有LIR基序,能够与自噬体ATG8家族蛋白相互作用,启动线粒体自噬。FBXL4也定位于线粒体外膜,介导BNIP3/BNIP3L的泛素化-蛋白酶体途径降解,使其维持本底低水平表达。而FBXL4缺乏则导致BNIP3/BNIP3L的大量积累,在培养的细胞系、MTDPS13患者的皮层神经元和FBXL4敲除/敲入小鼠模型中,FBXL4失活可触发高水平的基础线粒体自噬。FBXL4与其他外膜线粒体自噬受体,如PHB2、NIPSNAP1和FUNDC1没有相互作用。此外FBXL4缺乏诱导的线粒体自噬不依赖于经典的Parkin-PINK1途径。有趣的是,尽管FBXL4错义突变位点集中在负责和底物识别的LRR结构域,但这些突变并不影响FBXL4与BNIP3/BNIP3L的相互作用,而使其与复合体其他亚基如Skp1、Cullin1和RBX1的相互作用显著降低,出现活性CRL1FBXL4复合体的组装障碍,显著降低了对BNIP3/BNIP3L的泛素化降解作用。目前动态调控该过程的上游信号事件仍在探索中。
至今MTDPS13仍无有效治愈手段,支持性的临床干预措施功效甚微。近期的基础研究突破为未来探索该病的治疗策略提供了希望和思路。比如,在基于腺病毒的基因治疗方面,可将野生型FBXL4 cDNA引入携带FBXL4基因突变的患者细胞中或使用CRISPR碱基编辑来校正基因组DNA中的FBXL4突变;而非病毒的方法则可使用ASOs或RNAi降低BNIP3/BNIP3L的mRNA水平,从而缓解患者细胞中BNIP3/BNIP3L蛋白的异常积累;化学小分子也有望成为潜在的治疗策略,如设计PROTAC化合物靶向降解过量的BNIP3/BNIP3L;是否部分自噬抑制剂能够减轻患者细胞中过度的基础自噬也可以尝试;通过PGC-1α激动剂促进患者细胞中的线粒体生物发生(Mitochondrial biogenesis)以平衡过度的线粒体自噬可能也是一个潜在的探索途径。这些策略为MTDPS13未来治疗方法的开发提供了可能的方向,但需要进一步在临床转化模型中对其有效性和安全性进行系统检测和评估。
原文连接:https://doi.org/10.1016/j.molmed.2023.11.017