
溶酶体因其特有的60多种酸性水解酶,能够消化核酸、蛋白质、脂质及糖原等多种特定生物大分子, 该功能对于受损蛋白和细胞器的回收、能量和代谢稳态维持、以及细胞信号转导至关重要。越来越多的研究发现, 溶酶体功能异常导致一系列代谢相关遗传疾病,即溶酶体贮积症 (Lysosomal Storage Disease, LSD)。其中神经元蜡样脂褐质沉积症 (Neuronal Ceroid Lipofuscinoses, NCL)是LSD中数目最多的一类。该病的临床表现为渐进性失明、顽固性癫痫、进行性运动失调及早发夭折;病理表现为严重的脑萎缩、广泛的神经元变性丢失、以及自发荧光的脂褐质物质累积。NCL针对性的靶向治疗尚处于研究起步阶段。由于严重的临床症状和极其有限的治疗方式,NCL给病人、家属和社会带来了沉重的精神和经济负担。
目前共发现13个基因突变会导致NCL,这些基因大多编码溶酶体定位的水解酶类或负责溶酶体酶类从内质网向高尔基体、溶酶体转运的调节蛋白。KCTD7是最新鉴定到的NCL致病基因,编码一个Cullin 3家族E3泛素连接酶的底物识别亚基,但是其生理功能以及突变致病的分子机制尚不清楚。
2022年8月3日,复旦大学附属妇产科医院王红艳教授团队和生命科学学院王陈继副研究员团队合作在Science Advances在线发表了题为 KCTD7 mutations impair the trafficking of lysosomalenzymes through CLN5 accumulation to cause neuronalceroid lipofuscinoses的研究论文(图一)。该研究发现KCTD7失活导致CLN5蛋白异常积累在内质网,干扰了溶酶体水解酶类的分选和转运,进而导致溶酶体功能缺陷和疾病发生。

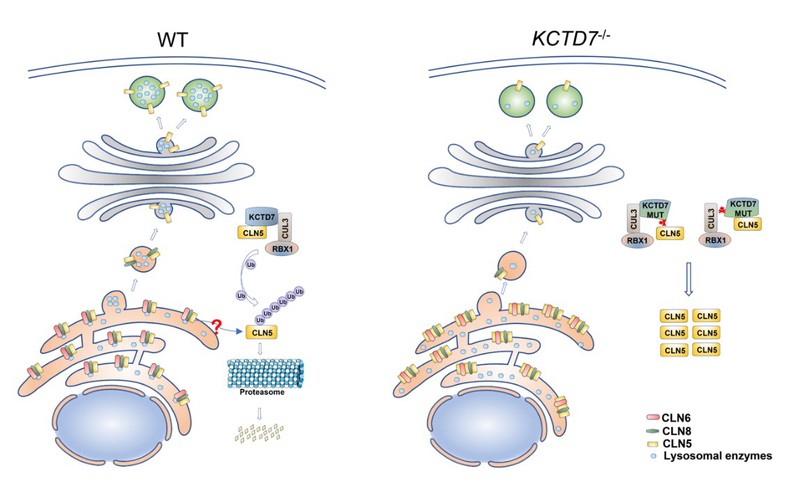
研究人员首先构建了KCTD7基因敲除的细胞株,发现KCTD7缺失细胞不仅可以重现病人所特有的电镜下深色嗜锇沉积物以及指纹样特征,而且还鉴定到了其他严重的溶酶体缺陷相关表型,包括中性脂质和糖原颗粒的异常积累、自噬缺陷、mTORC1信号下降、溶酶体水解酶类在溶酶体的定位效率下降、以及溶酶体相关的细胞死亡显著增加等。为了进一步探究KCTD7缺失导致上述表型的分子机制,研究人员纯化了内源KCTD7蛋白复合体,鉴定到CLN5是KCTD7的主要互作蛋白。值得注意的是,CLN5基因突变也会导致NCL的一个亚型。后续研究发现,KCTD7介导了CLN5的泛素化降解。NCL病人来源的KCTD7突变抑制KCTD7和CLN5的相互作用,或抑制其与CRL3复合体亚基Cul3的相互作用,从而破坏KCTD7对CLN5泛素化降解,导致CLN5在内质网异常累积。CLN5在正常情况下定位于内质网,协同促进CLN6/CLN8介导的溶酶体酶类互作和分选运输。但是在CLN5过量累积的情况下,CLN5/CLN6/CLN8复合体的化学计量(Stoichiometry)发生异常,继而导致溶酶体酶类从内质网到高尔基体的分选效率下降(图二)。
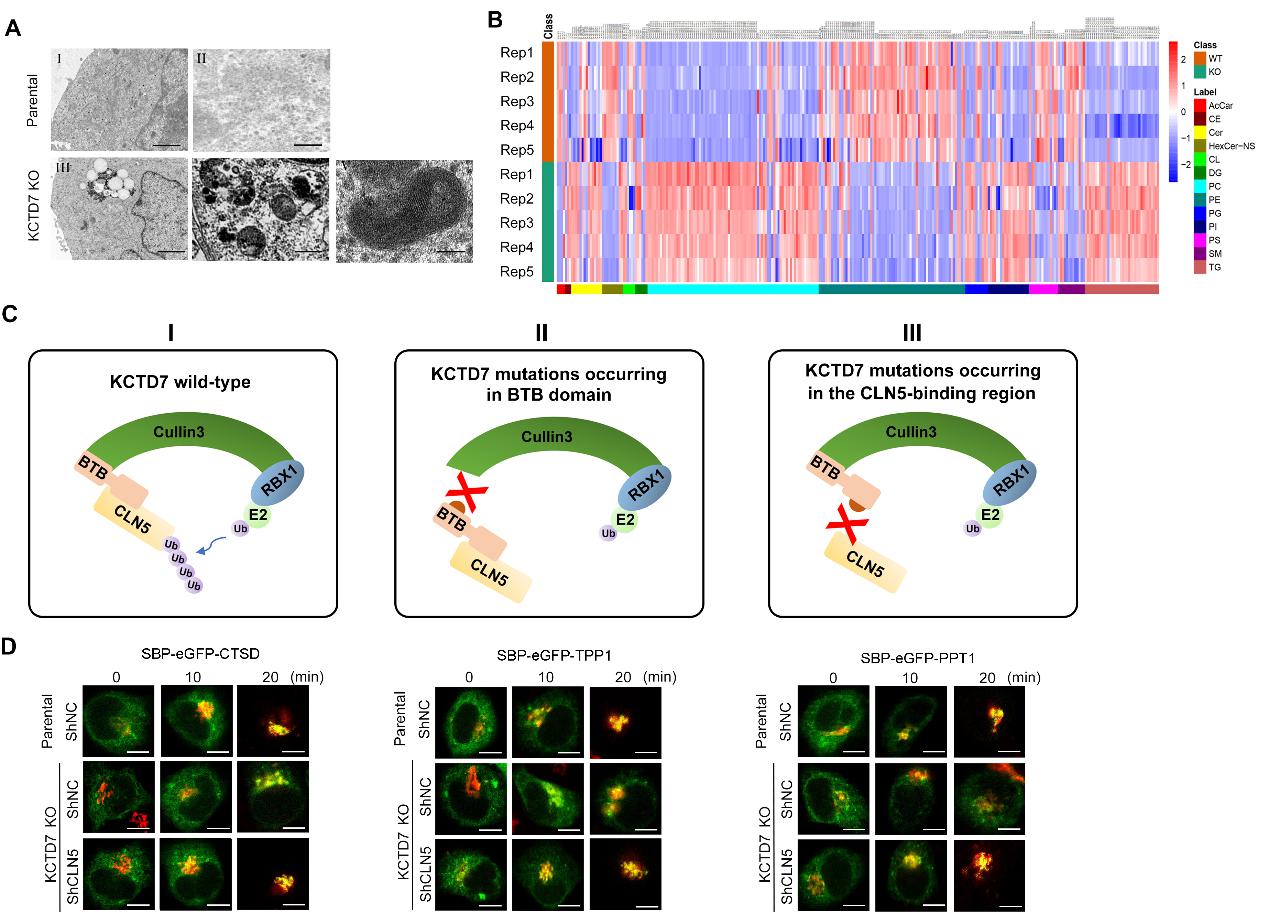
综上所述,研究人员揭示首次揭示了KCTD7在蛋白质稳态、溶酶体功能稳态维持中发挥关键作用,发现了KCTD7和CLN5这两种NCL相关蛋白在生化上相互关联,并解释了KCTD7突变的病理意义(图三)。该研究还提示,通过药物靶向降低CLN5蛋白水平是治疗KCTD7突变的NCL亚型的潜在靶向策略。
复旦大学附属妇产科医院博士后王亚兰为该论文第一作者。复旦大学附属妇产科医院王红艳教授和生命科学学院王陈继副研究员为该论文的通讯作者。
原文连接:https://doi.org/10.1126/sciadv.abm5578